Agonist and antagonist differences emerge for the β2 Adrenergic GPCR
Part 3 reports on work carried out up to January 2015 and continues from Part 1 (posted March 2014) and Part 2 (posted April 2014). Since January 2015, further work has revealed more information that is being written up for an academic paper.
The story so far
In Part 2 of this series, published online in April 2014, I described the ‘DRY’ lock mechanism and proposed that it was the best tool for monitoring the conversion of the β2AR from the active to inactive form. I also undertook to report how I would investigate the mechanism of the G-protein entry and exit from the active receptor.
However, “it is a truth universally acknowledged, that a scientific report distributed whilst relevant work is still in progress will be soon outdated!” My research is ongoing and my views have changed as a consequence. In reflection of this I have decided to continue reporting as I proceed.
Since posting Parts 1 and 2, I have done more work that showed that my initial results were neither consistent nor reproducible. I realize that this is because more preparatory care was needed on the input structures. I have carried out extensive work to address this problem and this work has revealed results that begin to suggest that we can distinguish agonists from antagonists and explain the way neutral antagonists work.
The continuing work also suggests that the ‘DRY’ lock may not be as important as I first thought, at least for the β2AR. Indeed, the definition of Asp130 and Arg131 (both on the receptor) as the accepted ‘DR’ part of the DRY lock is open to some confusion in the literature. I concluded that the Asp130/Arg131 ion pair is not appropriate as an indicator of active/passive receptor states and that a DR ion pair between Arg131 and Glu392 (on the G protein) may be more useful.
In this blog I report on the tentative finding that agonists stabilize the full complex and fight to keep the Arg131/Glu392 closed whilst antagonists destabilize the complex and encourage the ion pair to open and release the G-protein.
Making sure the input data set is sound
The majority of the effort involved in protein computation is to do with making sure the quality of the input structure is chemically sound. Most input data errors stem from incomplete and poorly refined X-ray coordinates.
Molecular mechanics can be very forgiving about yielding answers from poor structures but can fail if atom types are badly defined or bits of chemistry are missing. For example, a molecule of water and a piece of protein can be so close in the X-ray that they are inside a known physical barrier. Furthermore, along with other abstruse errors, many X-ray structures:
- Are missing entire residues
- Give a limited idea of which way protonic hydrogens may point
- Include the alpha carbons of named residues without the residue coordinates
- Have poorly defined terminations
- Cannot distinguish the orientation of terminal amide groups (ASN and GLN).
The most serious problems stem from the inability of most protein X-ray electron density maps to ‘see’ hydrogen atoms. Among other things, this means that the positions of hydrogen bonds, which are a major part of protein structure and action, have to be guessed, along with tautomeric states and formally chargeable ions. Group pKa values can vary drastically when imbedded in ‘protein space’ and distorting effects from neighbouring molecules in a crystal are rarely taken into account.
Where possible, correction routines have been developed to resolve these errors or at least highlight their presence. Plainly, entire missing residues cannot sensibly be replaced. General corrections to formal charge states can be applied with respect to the extent of solvation but pKa variations from known aqueous values are not yet tractable.
The general strategy for this work was to ‘clean’ the X-ray structure as far as possible and fully optimize every β2AR/G-protein (R*) complex using XED molecular mechanics before any analysis was attempted. Essentially, this strategy is a means of ‘normalizing’ all structures.
Molecular mechanics issues
Unlike quantum mechanics which is ab initio, molecular mechanics provides estimates of the strain in a structure based on empirical data from experimental observations. It returns a ‘strain energy’ (referred to as just the ‘energy’ in the bulk of this report). This ‘energy’ reflects how far the structure is from optimum but has no real physical counterpart.
Because entropic contributions are not included, the ‘strain energy’ should only be used for relative comparisons, e.g., the energies of two conformers of the same structure would be expected to have the same entropic contribution which, we assume, cancels on taking the energy difference. The same approximations apply to optimising structures by mathematically minimising their strain energy and comparing the difference in their final optimized energy.
The value of this ‘strain energy’ is unique to each molecular mechanics implementation. Since its usefulness rises with the quality of the molecular mechanics force field, I am keen to know if the XED force field can return valid conclusions from proteins and other large structures given its ability to take account of electron polarizability and improved handling of conjugated and aromatic systems.
The chosen ligands
A modified version of the table of chosen ligands from Part 2 is presented in Supplementary Material Appendix 1. Two main types of G-protein couple with the β2AR:
- Gi, which results in adenylate cyclase (AC) inhibition
- Gs, which stimulates it.
Current pharmacology strongly suggests the existence of at least three active β2AR* conformations that can either couple with both the Gi and Gs proteins or couple with one or the other.
The ‘action’ column in the table in Appendix 1 shows that propranolol inhibits Gs coupling without seemingly affecting Gi. Clenbuterol seems to agonise through Gi control only. ICI-118551 inhibits Gs and stimulates Gi. Since Gi stimulation acts as an AC controlling feedback mechanism, the stimulation of both Gi and Gs by some agonists and antagonists makes the agonist/antagonist distinction a matter of degree rather than definitive.
The main target for β2AR is the adenylate cyclase pathway. However, this versatile receptor stimulates other paths (MAPK, certain kinases and β-arrestin) but probably independently of the G-proteins and in some cases only after phosphorylation.
Improving the starting ligand conformations
As mentioned above, the early work could not be reproduced. This was apparent whenever small changes were applied to the conformation of the GR* complex or its ligand. The problem arises when minimising a large system with very many possible energy minima. Because minimisation merely moves energy downwards, the resulting optimized structure reflects the starting conformation. Therefore, if the starting conformation is wrong, the final structure will be wrong.
Simulated annealing techniques may overcome this problem somewhat, and dynamics should do even better, but the principle applies across the board, especially if the quality of the molecular mechanics is poor. It seemed reasonable to assume that more effort would be needed to get the starting conformations right.
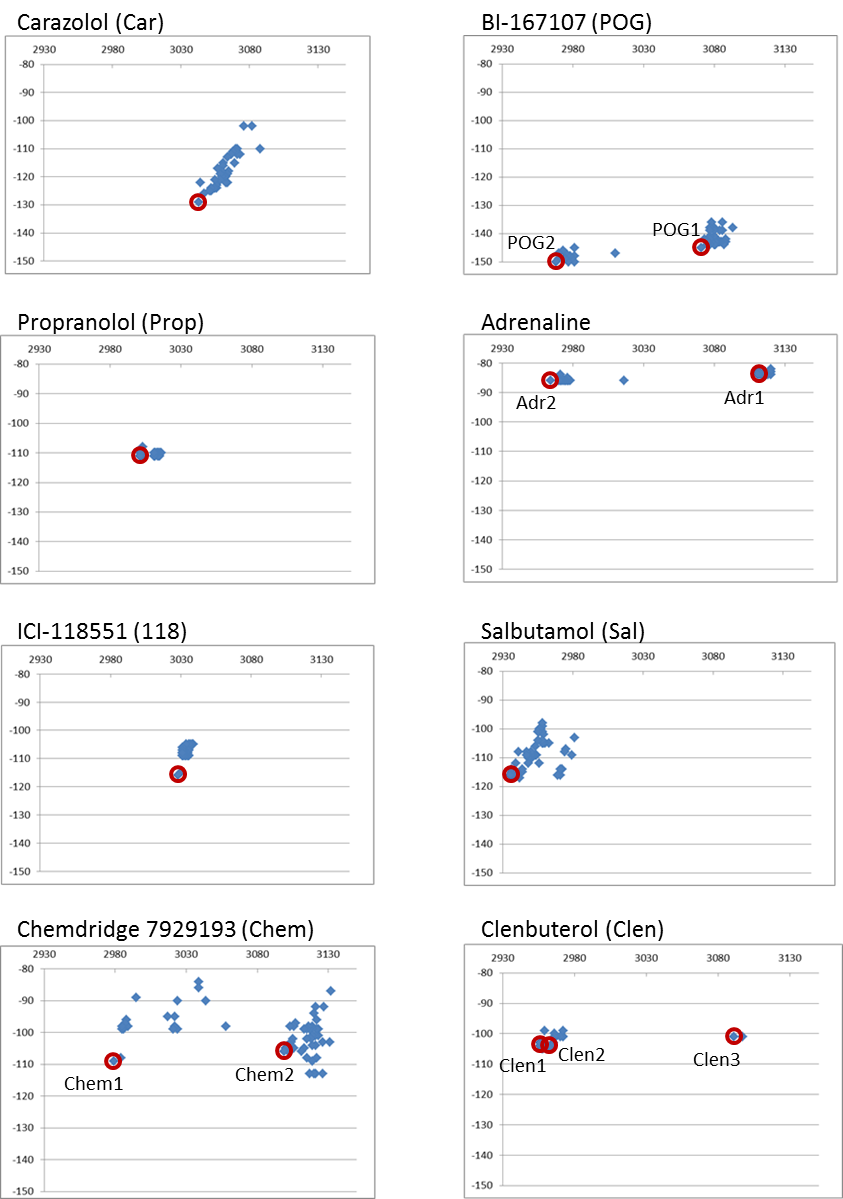
Between 45 and 80 randomly different conformations for each ligand were placed in the active site of the β2AR* crystal structure (truncated 3SN6.pdb coordinates) and the complete complex was minimized to a 0.02 rms exit limit. Supplementary Material Appendix 2 shows the plots of the binding energy of each ligand versus the total complex energy.
In principle, the conformation with the lowest total complex energy was deemed to be the desired starting conformation for each ligand. Using this criterion, new starting conformations for carazolol, propranolol, ICI-118551, and salbutamol were isolated from their conformational packs and placed into a ‘starting’ set.
However, two discrete conformational regions were identified for POG, adrenaline and clenbuterol and the lowest complex energy conformation was kept from each of the clusters. Two clusters were clearly discernible from the neutral antagonist ‘Chem 7929193’ (7929193-Chembridge) conformational collection but appeared much more loosely ordered than other ligands. Two relevant conformations were added to the starting set.
Analysing all minimized conformations
I now had a set of conformations of the fully optimized total complex (β2AR*+ligand) for each of the chosen ligands. The next question to ask was, are the conformers of this starting set related by structure?
Figure 7 summarizes the comparison of the Cα backbone of each conformer against all others. It may be inferred from this comparison that the inverse agonists share one unique conformation and the agonists share another.
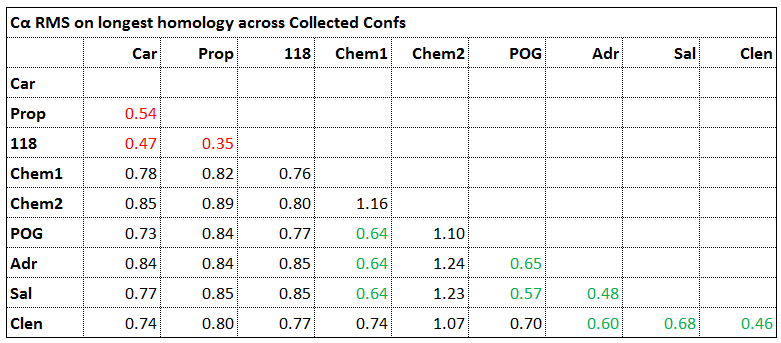
Figure 7 The Cα RMSD of each conformer across all others. The cut-off to discriminate ‘same’ conformers from ‘different’ conformers was taken as 0.7rms.
Figure 8 summarizes the analysis of these clusters with respect to their relative complex energies, the experimental pharmacology and the way each ligand affects the distance between the ‘DR’ (Arg131/Glu392) ion pair lock. This ‘DR’ lock (reported in Part 2) forms an ion pair between the G-protein and the receptor and is obviously going to be closed when the G-protein is bound. There may be variation in the other ion pairs that the two residues form after the G-protein leaves the receptor.

Figure 8. Summary of the analysis of Figure 7. The ligands in red are inverse agonists, those in green are agonists and ‘MT’ is the apo-complex (no ligand). The blue ligand is a neutral antagonist having no observable effect on the complex other than blocking its active site. Etot is the fully minimized complex energy for each ligand conformer started from the 3SN6.pdb truncated to include the G-protein surrogate and the receptor chains only. The estimated error attached to the full complex energy values is between ±10 and ±20 kcal/mole for approximately 6000 atoms and 6000 ‘xeds’ (polarisable extended electron constructs unique to XED molecular mechanics). The distance (Å) in column 3 is not the usual Arg131/Asp130, which shows little variation, but the alternative DR ‘lock’ across Arg131on the receptor and Glu392 on the G-protein.
The results shown in Figure 8 suggest that:
- The inverse agonists Carazolol, Propranolol and ICI-118551 destabilize the complex by ~20 kcal/mole over the empty complex and show the largest potential lock monitor distance
- The agonists POG, Adrenaline, Salbutamol and Clenbuterol decrease the energy over the empty complex by ~50 kcals/mole and show a tendency to decrease the potential lock monitor distance
- The neutral antagonist Chem 7929193 can stabilize two conformers with little change in binding energy, one of which decreases the stability over the empty complex by >100kcals/mole in line with inverse agonist action whilst the other stabilizes the complex by ~40kcal/mole in line with the agonists.
Chem 7929193 has been shown to be a neutral antagonist. Received wisdom, based on the ‘two state’ receptor theory, concludes that this category of antagonist rarely occurs as it necessitates agonism and inverse agonism to exactly cancel out. Our investigations of both the low and high energy forms of Chem 7929193 reveal conformers that seem to agonise and antagonise at the same time! Surely this must confuse the receptor and maybe bring it to a standstill whilst stopping any other ligand from entering the receptor.
In summary
If we assume that the conformations of the eight β2AR active complexes are correct, this work shows that there seems to be a relationship between the ligand type and its affect on G-protein/receptor complex stability. Why agonists appear to stabilize the complex more than inverse agonists is not yet clear. The ability of agonists to hold the Arg131/Asp130 DR lock more open than inverse agonists is intuitively attractive but inconsistently maintained across other GPCRs and unconfirmed in the present study.
Further work will concentrate on expanding the ligand base to see if the principle holds up, namely that G-protein/receptor stability is enhanced by agonists and diminished by inverse agonists.
Despite the lack of surrounding structure (membranes, water, sugars, etc.), the good correlation of the pharmacology of eight diverse β2AR* ligands with energies calculated using molecular mechanics is remarkable. Justification must reside in the fact that we are using a single system which is probably allowing cancellation of entropy, enthalpy, solvation and other neglected effects.
Supplementary Material
Appendix 1 – The β2AR ligands chosen for this study

Appendix 2 – Binding and Total Ligand Energies
Between 45 and 80 randomly different conformations for each ligand were placed in the active site of the β2AR crystal structure (truncated 3SN6.pdb coordinates) and the complete complex minimized to a 0.02 rms exit limit.